
Feb 16, 2023
Feb 16, 2023
We're Hiring
Join us and build the next generation AI stack - including silicon, hardware and software - the worldwide standard for AI compute
Join our teamTraining computer models for applications in computational chemistry can now be significantly accelerated thanks to the partnership between Graphcore and Pacific Northwest National Laboratory (PNNL).
The results of this partnership show that the training time for molecular graph neural networks (GNNs) can be drastically reduced by pretraining and finetuning the model when Graphcore intelligence processing units (IPUs) are used as an artificial intelligence (AI) accelerator.
These results were published online in Reducing Down(stream)time: Pretraining Molecular GNNs using Heterogeneous AI Accelerators1 and recently presented at the NeurIPS Workshop on Machine Learning and the Physical Sciences.
Molecular dynamics trajectories comparing pretrained and finetuned results from a graph neural network. Results show that the finetuned model does a much better job reproducing the ground truth method while remaining significantly faster to evaluate.
PNNL is a leading center for scientific discovery in chemistry, data analytics, Earth and life sciences, and for technological innovation in sustainable energy and national security.
By synergizing PNNL’s historic strength in chemistry with Graphcore’s advanced AI systems, the collaboration between the two enables the accelerated training of molecular GNNs.
The work thus far has focused on the SchNet GNN architecture2 applied to a large dataset—the HydroNet3 dataset—comprised of five million geometric structures of water clusters.
This GNN is designed for exploring the geometric structure – function relationship of molecules at a fraction of the computational cost of traditional computational chemistry methods.
Using the HydroNet dataset for this study was a natural fit—not only is it the largest collection of data for water cluster energetics reported to date, but its development was also spearheaded by PNNL researchers.
The SchNet model works by learning a mapping from a molecular structure to a quantum chemistry property. PNNL and Graphcore researchers are training the GNN to predict the binding energy of water clusters.
This energy is simply understood as a quantitative measure of how strongly the entire network of atoms is held together by chemical bonds.
The HydroNet dataset includes the binding energies which have been computed using highly accurate quantum-chemistry methods4.
The chemical bonds within a water cluster include a mixture of both strong covalent bonds with nearby atoms as well as long-range interactions through hydrogen bonding.
These interactions are known to be difficult to model in a performant manner. SchNet GNN neatly handles these by using a spatial cutoff to define pairwise interactions that grow linearly with the number of atoms being modelled and leveraging multiple message-passing steps to effectively propagate pairwise interactions across the entire structure to better incorporate the many-body nature of the bonding network.
Initial investigations into using SchNet for the HydroNet dataset have been limited to only using 10% of the total dataset5.
Even limiting the size of the dataset resulted in a time-to-train of 2.7 days with a reported time-per-epoch of 4.5 minutes using four NVIDIA V100 GPUs.
The graph below shows the speedup for the time-per-epoch by using Graphcore accelerated training of SchNet over the same small subset (10%) of the HydroNet dataset.
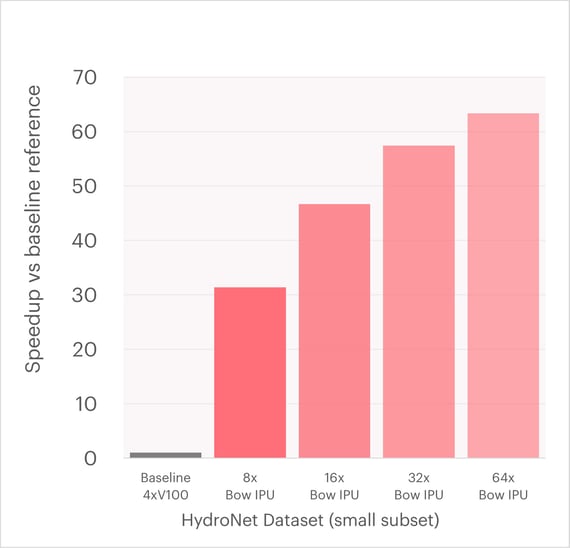
Speedup (higher is better) over baseline implementation using 4xV100 NVIDIA GPUs1. Over 60x acceleration is possible by using 64 IPUs over the small subset of 500k water clusters2.
These results show that a Graphcore Bow Pod16 achieves an over 40x speedup over the previous baseline implementation using legacy hardware accelerators.
Putting these results into perspective, using published pricing for Paperspace gradient machines, the 2.7 days training time on 4xV100 GPUs would cost $596. By contrast, on the Bow Pod16 the same training workload would take just 1.4 hours and cost $37.
These comparisons are just for the small subset of the HydroNet dataset. The cost and model development time savings are amplified when considering scaling to training over larger datasets as well as sweeping over different hyperparameters.
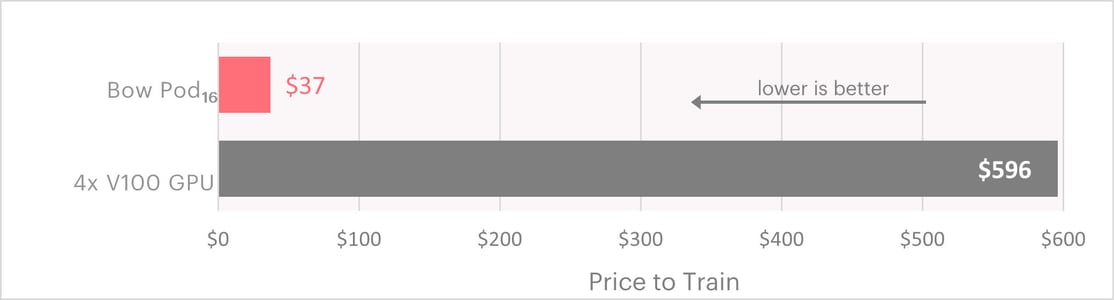
Computing cost comparison for training SchNet GNN on HydroNet sataset using Paperspace cloud services
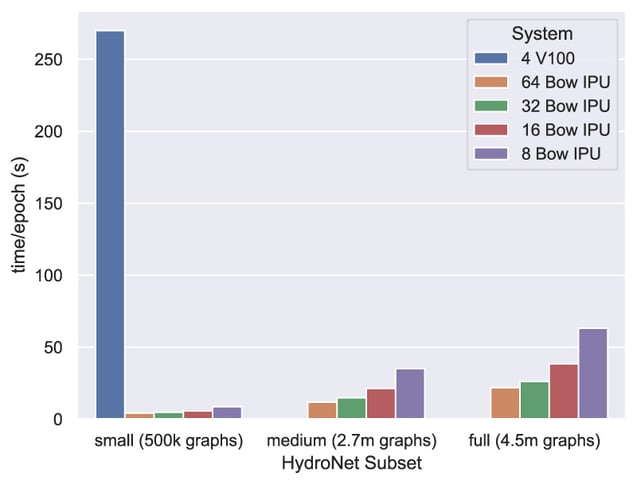
Time per epoch (lower is better) collected over a range of Bow IPU systems compared with the baseline implementation.
More importantly, by using the Graphcore Pod systems our collaboration with PNNL has demonstrated efficient training over the full dataset. The features and capabilities of the Graphcore IPU that make this possible are explored in greater detail in Extreme Acceleration of Graph Neural Network-based Prediction Models for Quantum Chemistry6.
This effort is just one small but significant step towards using machine learning to further the understanding of complex many-body interactions at the atomic scale and how those relate to macroscopic observables. Using these methods can enable breakthroughs across a range of applications where understanding of chemical bonding is essential—from solar panels and batteries to drug discovery.
1. Bilbrey, J. A. et al. Reducing Down(stream)time: Pretraining Molecular GNNs using Heterogeneous AI Accelerators. Preprint at https://arxiv.org/abs/2211.04598 (2022).
2. Schütt, K. T., Sauceda, H. E., Kindermans, P. J., Tkatchenko, A. & Müller, K. R. SchNet - A deep learning architecture for molecules and materials. J. Chem. Phys. 148, 1–11 (2018).
3. Choudhury, S. et al. HydroNet: Benchmark Tasks for Preserving Intermolecular Interactions and Structural Motifs in Predictive and Generative Models for Molecular Data. Preprint at https://arxiv.org/abs/2012.00131 (2020).
4. Rakshit, A., Bandyopadhyay, P., Heindel, J. P. & Xantheas, S. S. Atlas of putative minima and low-lying energy networks of water clusters n = 3-25. J. Chem. Phys. 151, (2019).
5. Bilbrey, J. A. et al. A look inside the black box: Using graph-theoretical descriptors to interpret a Continuous-Filter Convolutional Neural Network (CF-CNN) trained on the global and local minimum energy structures of neutral water clusters. J. Chem. Phys. 153, (2020).
6. Helal, H. et al. Extreme Acceleration of Graph Neural Network-based Prediction Models for Quantum Chemistry. Preprint at https://arxiv.org/abs/2211.13853 (2022).
Note: Cover image is a visualisation of molecules created using Midjourney AI and is not intended to represent the work described in this blog.
Share:


